(6/8)
2-2-3.新しい相同性検索法 PSI-BLAST
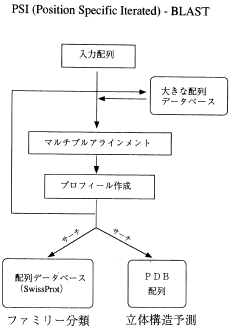
画面22
画面23
そのうちに、とんでもない方向から、「PSI-BLAST」という非常に優秀な予測法が出てきました。PSI-BLASTとは、既に世界中で使われていたホモロジー検索法「BLAST」の拡張版であり、同一の開発者によって1997年に発表された方法ですが、これはあくまでホモロジー検索のために開発した方法であり、構造予測のためのものではありませんでした。しかし、これを予測法に用いると、3D-1Dと同じように非常に弱いレベルのホモロジーまで検知し、かつ、3D-1Dに比べ性能が非常によく、信頼性が高いという性質をもっていたのです。
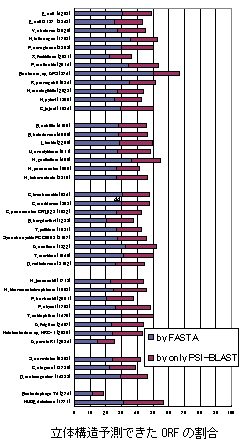
それでは、何がそれほど性能をよくしているかといいますと、従来のBLASTは問合わせ配列(query) を使ってそのまま配列サーチするのに対し、PSI-BLASTの場合は、queryのホモロジー配列の情報も加えた非常に大きなデータベースにかけ、似たものを全て引っぱってくるためです。つまり、まずホモロジー配列の「マルチプルアラインメント」を作り、全ての情報をプロファイル形式にまとめ〜この点は3D-1Dのプロファイルと似たような形式ですが〜、このプロファイルを用いてデータベースに対しサーチするわけです。ホモロジー検索の場合には配列データベースをサーチさせますが、構造予測をしたい場合には、PDB(Protein Data Bank)のデータベースを検索させます。PSI-BLASTはあくまで配列どうしを比較するホモロジー検索法ですが、配列が似ていれば構造も似ているであろうという論理により、構造予測法としても活用できるわけです。折りしもゲノムの時代が始まり、我々をはじめ多くの研究者が、PSI-BLASTをゲノムに応用するとどのくらいの威力があるかを検討しはじめました。画面24は、実際に我々が計測した結果ですが、大腸菌やバクテリア、ショウジョウバエなど、全ゲノム中の遺伝子の立体構造を、FASTA (BLASTと同様に当時広く使われていた相同性検索プログラム)、PSI-BLASTのそれぞれで予測を行った結果です。それぞれの生物種は4000〜20000個以上の数の遺伝子をもっていますが、全体を100とした場合の割合(%)を単純に比較すると、PSI-BLASTが、全体の45%の立体構造を予測できるという結果がでました。我々は、これらの解析結果を、すべて「GTOP」というデータベースに登録し、WEBにて公開しています(http://spock.genes.nig.ac.jp/~genome/gtop.html)。
以上が、これまでのおおよその研究史です。このように、PSI-BLASTは実用的に非常によい方法ですが、それでもやはり45%しか予測できないという限界があるわけです。そこで、現在注目されている方法である「アブイニシオ法」についてご説明しましょう。
画面24
画面25